Jul 29 2020
Quantum computers exhibit tremendous potential for computations with the help of innovative algorithms and handle huge amounts of data much higher than the capacity of existing supercomputers.
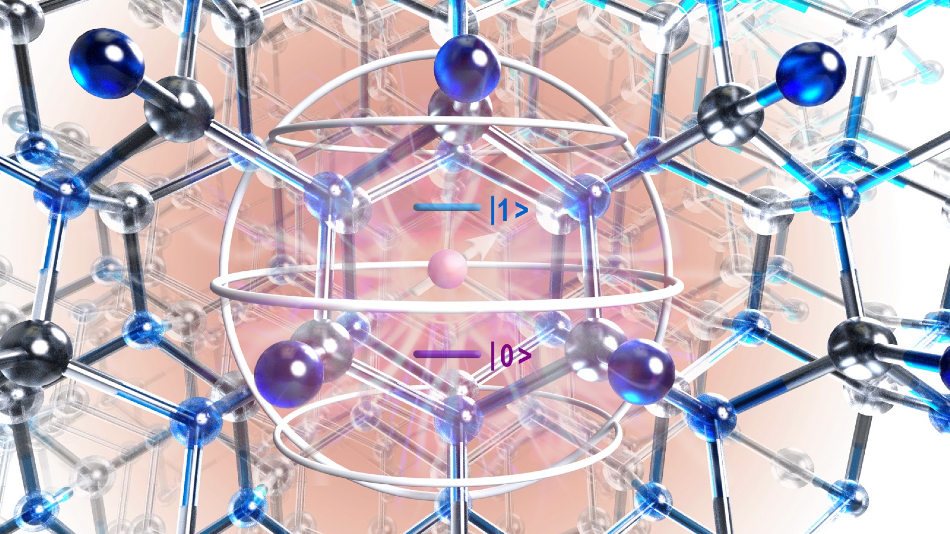 Artistic rendering of the atomic structure of silicon carbide crystal showing defect (purple circle) and region of interest identified with quantum mechanical theory (silver sphere). Image Credit: University of Chicago.
Artistic rendering of the atomic structure of silicon carbide crystal showing defect (purple circle) and region of interest identified with quantum mechanical theory (silver sphere). Image Credit: University of Chicago.
Although such quantum computers have already been developed, they are still in their earlier and face limited applicability to solve complex problems in chemistry and materials science. For instance, they only enable the properties of a few atoms to be simulated for materials research.
Researchers from the U.S. Department of Energy’s (DOE) Argonne National Laboratory and the University of Chicago (UChicago) have created a new technique that enables the use of quantum computers to simulate complex materials and realistic molecules, the description of which necessitates hundreds of atoms.
The team of researchers is headed by Giulia Galli, director of the Midwest Integrated Center for Computational Materials (MICCoM), a group leader in Argonne’s Materials Science division and a member of the Center for Molecular Engineering at Argonne.
Galli is also the Liew Family Professor of Electronic Structure and Simulations in the Pritzker School of Molecular Engineering and a Professor of Chemistry at UChicago. She contributed to this study along with assistant scientist Marco Govoni and graduate student He Ma, both part of Argonne’s Materials Science division and UChicago.
Our newly developed calculational method. greatly improves on the accuracy attainable with existing quantum mechanical methods regarding calculations for specific defects in crystalline materials, and we have implemented it on a quantum computer.
Giulia Galli, Director, Midwest Integrated Center for Computational Materials
Over the past 30 years, theoretical approaches based on quantum mechanics have been crucial in estimating the properties of materials applicable to quantum information science, as well as functional materials for energy storage systems, energy applications, and encompassing catalysts.
But these approaches are computationally challenging, and it is still difficult to use them in the case of complex, heterogeneous materials.
In our research we developed a quantum embedding theory that permitted the simulation of ‘spin defects’ in solids by coupling quantum and classical computing hardware.
Marco Govoni, Assistant Scientist, Materials Science Division and Center for Molecular Engineering, Argonne National Laboratory
Such defects in solids can be leveraged to develop materials relevant to quantum information processing and nanoscale sensing applications that exceed existing capabilities.
“Ours is a powerful forward-looking strategy in computational materials science with the potential of predicting the properties of complex materials more accurately than the most advanced current methods can do at present,” added Govoni.
Initially, the researchers tested the quantum embedding approach on a classical computer, using it to estimate the properties of spin defects in silicon carbide and diamond.
Past researchers have extensively studied defects in both diamond and silicon carbide, so we had abundant experimental data to compare with our method’s predictions.
He Ma, Graduate Student, Materials Science Division and Center for Molecular Engineering, Argonne National Laboratory
With the ideal agreement between experiment and theory, the researchers were confident in the reliability of their method.
Then, they endeavored to test the same calculations on a quantum simulator and ultimately on the IBM Q5 Yorktown quantum computer. The outcomes confirmed the effectiveness and high accuracy of their quantum embedding approach, thus offering a novel platform to help solve several different types of materials science problems using a quantum computer.
With the inevitable maturity of quantum computers, we expect our approach will be applicable to the simulation of regions of interest in molecules and materials for the understanding and discovery of catalysts and new drugs, as well as aqueous solutions containing complex dissolved species.
Giulia Galli, Director, Midwest Integrated Center for Computational Materials
Galli’s research team is part of MICCoM, headquartered at Argonne; the Chicago Quantum Exchange, headquartered at UChicago; and the QISpin project financially supported by the Air Force Office of Scientific Research.
The study employed the WEST software created within MICCoM and used various computing resources apart from the publicly available IBM quantum computer: the Argonne Leadership Computing Facility and the National Energy Research Scientific Computing Center, both DOE Office of Science User Facilities; and the University of Chicago Research Computing Center.
The study was financially supported by the DOE Office of Science and the Air Force Office of Scientific Research.
Journal Reference:
Ma, H., et al. (2020) Quantum simulations of materials on near-term quantum computers. npj Computational Materials. doi.org/10.1038/s41524-020-00353-z.